Dorado v2.0.0 part 2: assembly polishing
~•

![]()
This post follows my previous post on Dorado v2. I’ll skip background and assembly methods here, so you may want to read that one first.
The key finding in my last post was that the new hac@v6.0.0 basecalling model is not as good as the older sup@v5.2.0 model, with both read accuracy and assembly accuracy. However, dorado polish has the potential to fix assembly errors. If polished assemblies of hac@v6.0.0 reads are as good as (or better than) polished assemblies of sup@v5.2.0 reads, that could change my mind about the new basecalling model.
Polishing models
Dorado’s polishing models seem to come in two flavours: older pileup models and newer read-level models. The pileup models work from a summary of the bases seen at each assembly position. They have a simpler architecture: two 128-dimensional bidirectional GRU layers, 405k total parameters. The read-level models retain per-read information instead of collapsing everything into a pileup. They are larger: a CNN followed by four 384-dimensional LSTM layers, 4.85m total parameters, so ~12× larger than the pileup models.
Each of the basecalling models I tested has two read-level models: one that does not use move tables and one that does:
dna_r10.4.1_e8.2_400bps_hac@v5.2.0_polish_rldna_r10.4.1_e8.2_400bps_hac@v5.2.0_polish_rl_mvdna_r10.4.1_e8.2_400bps_hac@v6.0.0_polish_rldna_r10.4.1_e8.2_400bps_hac@v6.0.0_polish_rl_mvdna_r10.4.1_e8.2_400bps_sup@v5.2.0_polish_rldna_r10.4.1_e8.2_400bps_sup@v5.2.0_polish_rl_mv
In these model names, rl appears to stand for ‘read-level’ and mv stands for ‘move table’. Move tables contain information on how the basecalled sequence aligns to the raw signal and therefore how much signal is associated with each base – an extra layer of information which is potentially useful for polishing. To get move tables, you need to use --emit-moves when basecalling.
I also checked the actual weights in these read-level models, and they are all different. E.g. while they have the same architecture, the hac@v5.2.0_polish_rl, hac@v6.0.0_polish_rl and sup@v5.2.0_polish_rl models all appear to have been trained separately.
In addition to those basecalling-model-specific polishing models, there is a pileup bacterial model named dna_r10.4.1_e8.2_400bps_polish_bacterial_methylation_v5.0.0. This model is different in that it spans many basecalling models – despite the v5.0.0 in the model name, it is not just for v5.0.0 basecalling models. As you can see in the Dorado source code, dorado polish --bacteria will use this model for hac and sup basecallers from v4.2.0 to v6.0.0.
So for each of the three basecalling models I used, there are three possible polishing models to test: bacterial, RL (read-level without move tables) and RL+moves (read-level with move tables).1
Methods
I started with 150 assemblies: 5 genomes × 3 basecalling models × 10 read subsets. I excluded 3 of these (Shigella assemblies from read subset 8), leaving 147.2 I then polished each assembly using dorado polish with the three possible polishing models: bacterial, RL and RL+moves. The bacterial model was the same for all assemblies, while the RL and RL+moves models depended on the basecalling model. This resulted in 441 polished assemblies.
I analysed these using the same assembly analysis script as before. I also did an analysis on each polishing change, classifying as an improvement (making the assembly closer to the reference), a degradation (making the assembly further from the reference) or a neutral change (not changing the assembly-to-reference distance, e.g. changing an incorrect base to a different incorrect base).
Speed performance
The last time I tested polishing, I ran dorado polish on the CPU and found that the smaller bacterial model took a few minutes to run while the larger RL models took about an hour. Since I had hundreds of assemblies to polish this time, I ran dorado polish on an H100 GPU (via University of Melbourne’s Spartan cluster), and it was very quick. The bacterial model typically took 4-5 seconds per assembly while the RL models took 10-12 seconds – hundreds of times faster than on CPU.
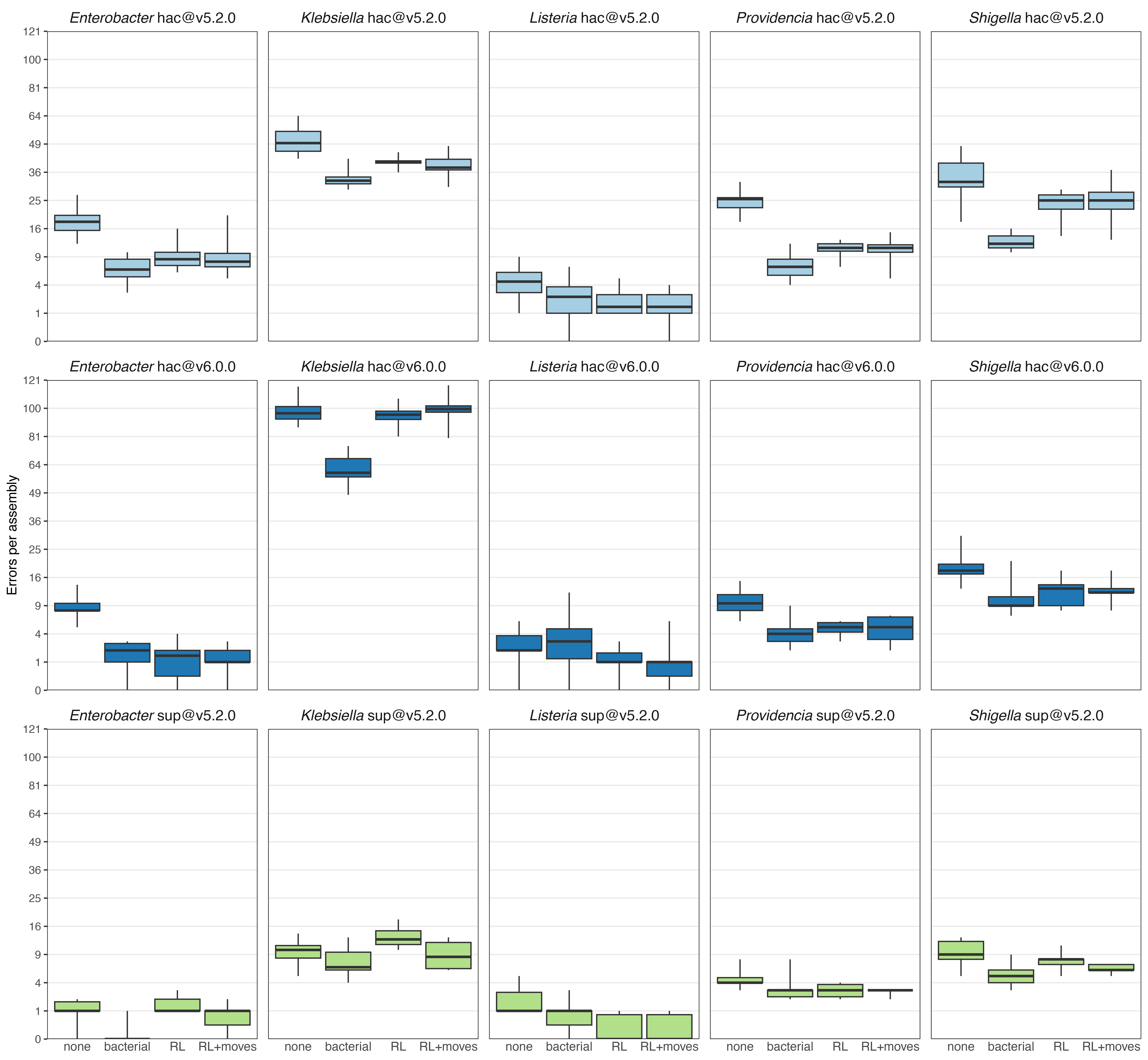
Polished assembly accuracy
The below plot contains the main results, showing assembly errors (lower is better) on the y-axis. The ‘none’ values contain no polishing and match the assembly boxplot in the previous post.

It is clear that polishing tends to make assemblies better on average – the median errors per assembly dropped for every polishing model and every basecalling model. The hac@v6.0.0 polished assemblies are pretty good, but the sup@v5.2.0 polished assemblies are still better.
But there were cases where assemblies got worse with polishing. The table below shows totals at the per-assembly level:
| Polishing model | Worse | Same | Better |
|---|---|---|---|
| bacterial | 6 | 12 | 129 |
| RL | 22 | 10 | 115 |
| RL+moves | 14 | 16 | 117 |
The same pattern appears when looking at individual polishing changes. Most polishing changes were improvements (removed an error), but some polishing changes were degradations (added an error):
| Polishing model | Degradations | Neutral | Improvements |
|---|---|---|---|
| bacterial | 300 | 18 | 1615 |
| RL | 604 | 20 | 1275 |
| RL+moves | 587 | 21 | 1288 |
Klebsiella
As I saw in the last post, when the results are separated by species, the Klebsiella genome stands out: it has far more errors than the other genomes, and polishing with the read-level models produced little or no improvement in assembly quality (though the bacterial model still did).
{kind=link}
Mike Vella (VP of Machine Learning at Oxford Nanopore) emailed me with a possible explanation: these errors could be due to phosphorothioate modifications which were absent from their latest training run. This is where an oxygen atom in the DNA backbone is replaced with sulfur.3 While not a base modification, it can presumably still affect the raw ONT signal (the squiggle) and therefore impact basecalling. Mike let me know that ONT is working on an updated dorado polish model to correct this, so I’ll keep an eye out for that.4
9 July 2026 update: As I said in the footnote, I didn’t find a dnd gene cluster in this genome, so phosphorothioate mods were never a great fit. But Michael Biggel (University of Zurich) emailed me and suggested I look for the dpd system which does 7-deazaguanine mods, and this was indeed in the Klebsiella genome. So I think this mystery is now solved: 7-deazaguanine modifications from the dpd system are causing the higher error rate. Thanks to Michael for figuring this one out and letting me know. Here’s a link to his preprint which covers this: Standalone nanopore sequencing for foodborne pathogen surveillance: a large-scale evaluation and quality control framework.
Discussion and conclusions
In this test, polishing usually improved assembly accuracy, but not enough to close the gap between hac@v6.0.0 and sup@v5.2.0. So my recommendation from the last post still stands: stick with sup@v5.2.0 basecalling.
Comparing polishing models, the bacterial model did best, despite being older, smaller, not tied to a specific basecalling model and not using move tables. I therefore feel comfortable with this recommendation: if you have an ONT-only assembly of a bacterial genome, run dorado polish --bacteria.
But why did the older, smaller bacterial model outperform the newer read-level models, even those using move tables? I hypothesise it may be down to the training data. A move-table-aware read-level model has more parameters and richer input data, so it should be able to outperform the simpler pileup model, but only with sufficiently diverse bacterial training data. It would be great to see ONT train bacterial-specific read-level models, or at least add more native bacterial DNA to the training data for their general polishing models.
Even though polishing usually helps overall, the fact that it can introduce new errors is concerning. In this post, I used dorado polish to directly produce polished FASTA files. A more controlled approach would be to run it with --vcf, filter the proposed variants and then only apply the high-confidence ones. If the variants that create errors are distinguishable by quality score, depth or any other metric, this could improve polishing precision. Something to try!
Finally, here are some tips on how I curate polishing on an ONT-only assembly:
- This script shows polishing changes in a human-readable format. With sup-basecalled Autocycler assemblies, there are often few enough changes to review them manually.
- If
dorado polishmakes many changes clustered in one region, that’s suspicious and I usually discard the changes. I describe one such case in this post. - For indel changes, I compare mean Prodigal length before and after the change. Longer predicted proteins suggest the indel fixed a frameshift error, while shorter ones suggest it may have created one.
- I can also look for exact matches to public genomes. If the polished version is common and the unpolished version is absent, I am more inclined to believe the change. However, this makes the polishing biased toward known sequences and no longer 100% de novo.
Footnotes
-
The state of
dorado polishmodels is somewhat confusing. Some polishing models have big architectures, some are small. Some use move tables (and so require BAM reads produced withdorado basecaller --emit-moves), some don’t. Some have bacterial-specific training, some are general. Some are tied to a specific basecalling model, some span many models. ↩ -
Autocycler did not give complete assemblies for these three because they had a region with very low read depth (down to 1× at some positions). To produce the complete assemblies, I patched them together using the reference genome as a guide, and this was fine for the last blog post where I was just concerned with small-scale errors in the assembly. But for this post, it doesn’t make sense to polish regions that wouldn’t assemble, so I excluded those assemblies. ↩
-
I must confess that I was not familiar with this modification until I got Mike’s email. Here’s an interesting paper that I found helpful: The origin and impeded dissemination of the DNA phosphorothioation system in prokaryotes. However, I looked for a dnd or ssp gene cluster (would be strong evidence for phosphorothioate mods) in the Klebsiella genome and did not find anything, so I’m still open to other explanations for the Klebsiella errors. ↩
-
While ONT is planning a new polishing model to address this particular issue, it would be nice to also see a basecalling model trained on phosphorothioate modifications to fix the problem one step earlier. ↩