Medaka v2: progress and potential pitfalls
•

![]()
A new version of Medaka was recently released, featuring a model designed specifically for bacterial genomes. See this video and this video from ONT for an overview of the challenges posed by modified bases and some information on the new Medaka release.
As someone focused on bacterial genome assembly, this piqued my interest! Over the past few years, I’ve mostly moved away from using Medaka, as it didn’t usually improve assemblies from sup-basecalled reads.1 Could this new model change things? In this post, I’ll share my initial thoughts on the new Medaka model and highlight a key pitfall in genome polishing.
New Medaka model
In Medaka v2.0.0, you can specify the r1041_e82_400bps_bacterial_methylation model or simply use the --bacteria flag. This model is flexible with basecalling, supporting v4.2, v4.3 and v5.0 basecalls at both hac and sup speeds.2
To quickly test it, I used two genomes with higher-than-normal error rates in my ONT-only assembly: a Campylobacter lari genome with 18 errors and an Enterobacter cloacae genome with 11 errors. While these numbers may seem low, most of my ONT-only assemblies in 2024 have fewer than five errors. I suspect methylation motifs are behind these higher error rates.
| Genome | Pre-Medaka errors | Medaka v1.12.1 errors | Medaka v2.0.0 errors |
|---|---|---|---|
| C. lari | 18 | 28 | 2 |
| E. cloacae | 11 | 30 | 10 |
In both cases, the previous release of Medaka made the accuracy worse and the new release made it better! While this is based on just two genomes, it’s enough to reignite my interest in Medaka.
Missing plasmid pitfall
Encouraged by these results, I ran the new Medaka model on some Staphylococcus aureus assemblies from a current project. But to my surprise, Medaka often made the assemblies worse.
This genome has a 2.9 Mbp chromosome and two small plasmids: 4.4 kbp and 3.1 kbp. As I dug into the strange Medaka results, I found that the chromosome and 4.4 kbp plasmid share ~850 bp of sequence with ~80% identity. This normally wouldn’t be a problem, but some assemblies were missing that plasmid.3
Here’s an IGV screenshot of the problematic region in Medaka’s calls_to_draft.bam file from one such assembly:
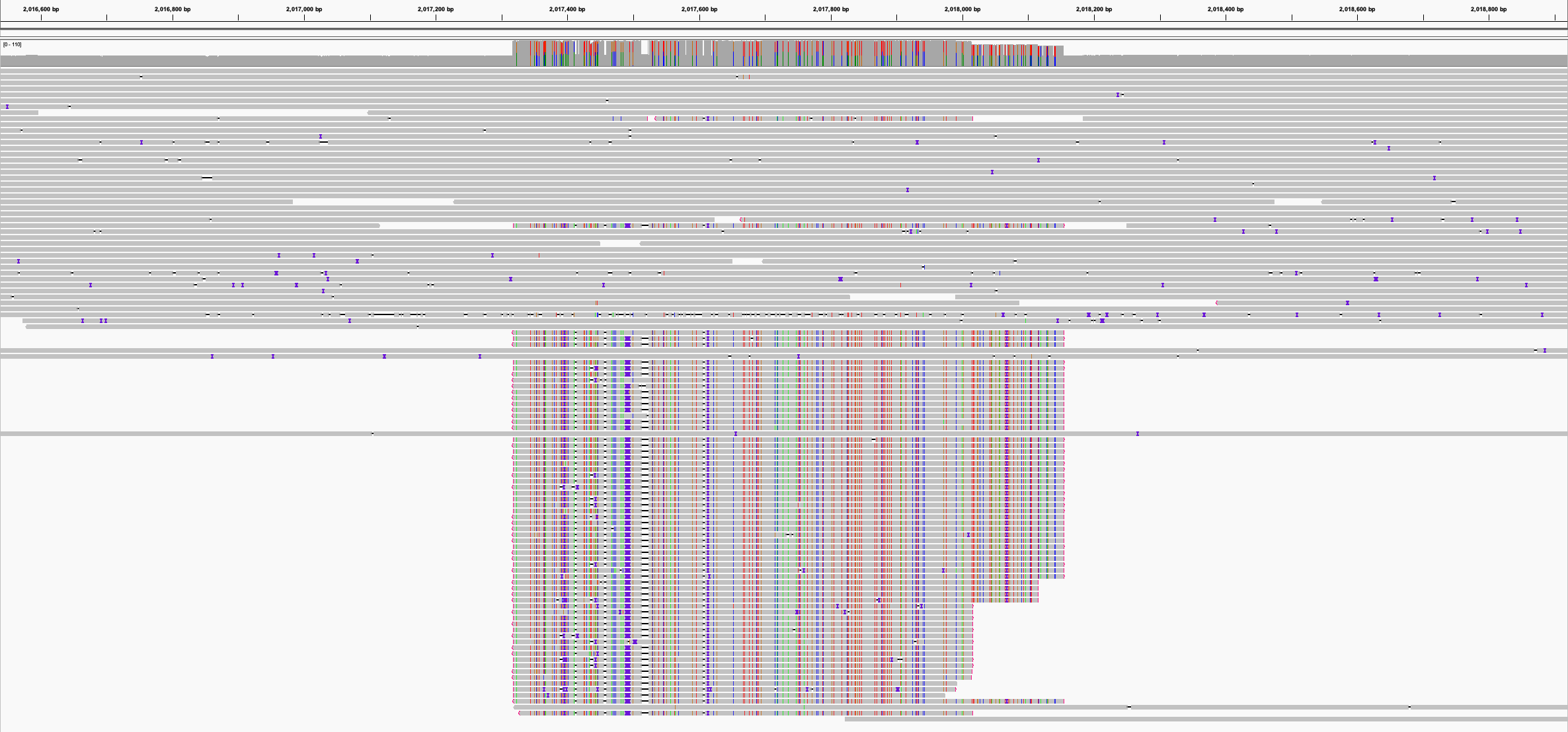
In the screenshot, the reads that align cleanly are from the chromosome, but others (at the bottom) are plasmid reads that erroneously aligned to the chromosome since the plasmid was missing. The plasmid reads outnumbered the correct chromosome reads, leading Medaka to introduce over 100 changes – all of them errors.
Lesson learned: Make sure your assembly is structurally sound before running Medaka. Missing plasmids can cause havoc during polishing.4 I also noticed that circularisation overlap (duplicated sequence at contig ends) sometimes acquired errors during Medaka polishing.5
To avoid this problem, I recommend trying Hybracter from George Bouras. It includes special logic to recover small plasmids which may be missing from the long-read assembly.6 Alternatively, you could filter out any reads that don’t have a high-identity full-length alignment before running Medaka.
Final thoughts
While I am impressed by the new version of Medaka, if you have short reads available, you can probably skip it and go straight to polishing with Polypolish and/or Pypolca. But if you don’t have short reads, the new Medaka model is worth a try.
After running Medaka, how can you assess whether the polished assembly is better or worse? There’s no perfect solution, but I find the mean length of predicted proteins to be a decent metric.7 It’s also good practice to manually inspect Medaka’s changes.8 Ideally, Medaka should make scattered, minimal changes. If you notice clusters of changes in a single region, that’s a red flag – not just for Medaka, but for any polisher.
Footnotes
-
Other Medaka models are specific to a basecalling version and speed, e.g.
r1041_e82_400bps_sup_v5.0.0. Presumably this reflects the data that model was trained on, so the new bacterial Medaka model was probably trained on a mix of different basecalling models. ↩ -
A missing small plasmid is a common problem with long-read assemblies. Sometimes it’s due to library-prep bias (see this paper), but it can also be a fault of the assembler. ↩
-
This missing plasmid issue isn’t specific to the new Medaka version – earlier versions would have encountered the same problem. ↩
-
Canu contigs usually have circularisation overlap, but the contig headers specify how much should be trimmed, so I wrote this script to automate the trimming. I suspect Canu assemblies cleaned by this script will perform better in Medaka than unprocessed Canu assemblies. ↩
-
Read more about Hybracter in this paper and this blog post. ↩
-
Assembly errors can truncate coding sequences, so assemblies with more errors tend to have shorter predicted proteins. So when comparing alternative assemblies, the one with a larger mean protein length is likely better. This metric is more sensitive to indel errors than substitutions, because indels create frameshifts which often lead to premature stop codons. A helper script to measure this is available here. ↩
-
I wrote this script to display pre- and post-polishing differences in a human-readable format. ↩